| Home | |
| Genome-Phenome Analyzer |
Recently added capabilities in the Genome-Phenome Analyzer
Common variants that appear to be de novo are filtered out by default (22 Aug 2017)
If a variant is read as de novo and is more common than the frequency that would account for all diseases known to have this gene zygosity, the default for the software is now for the variant to be filtered out. The number of variants filtered in this way is reported along with other outputs of the variant file reading.
This default can set as desired for each institution, or can be changed in any analysis by changing the following setting on the Set Variant Parameters screen:
![]()
This uses information displayed in light blue on the mini variant tables (see Frequency context for variants below) and filters out the variants in circumstances in which they are not plausible.
Pseudoautosomal gene list augmented with non-protein-producing genes (4 May 2017)
Although there are no known genetic disorders involving non-protein-producing pseudoautosomal genes, these are now added to the software's list of pseudoautosomal genes to prevent unnecessary flagging of gene variants inconsistent with sex.
Variants with poor reads or depth are flagged (23 March 2017)
A variant that has a poor depth or quality score in the proband is filtered out of the analysis in the SimulConsult Genome-Phenome Analyzer. However, if the proban's scores are acceptable and parental scores are not, the variant is displayed anyway. To draw attention to the questionable zygosity for the parents in such situations, the parent’s zygosity is now flagged with a question mark, as illustrated below. As indicated in the key, hovering over the zygosity gives the details of depth and quality:

Transition to ACMG "Secondary" (Incidental) Findings version 2.0 (October 2016)
As detailed in the recommendations, 4 genes were added and one removed.
Speeding up of flagging of alternative reads (27 June 2016)
For variant tables with alternative reads on different row it is necessary to flag alternative reads to avoid creating compound heterozygotes from the alternative reads. This was the case only for a small minority of users, but the necessary flagging added hugely to the processing times for large variant tables.
The software now uses a more efficient algorithm that reduces the time for this flagging to the point where there is no noticible delay, resulting in the entire analysis taking ~2 seconds for ~100,000 variants.
Frequency headers are reflected in the mini variant table (6 June 2016)
For the case shown below, the two frequency columns had headers freq1000 and freqLocal, and the identifiers were parsed to produce the following headers in the mini variant table shown below: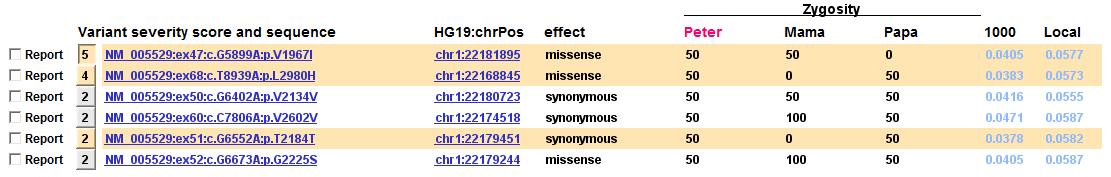
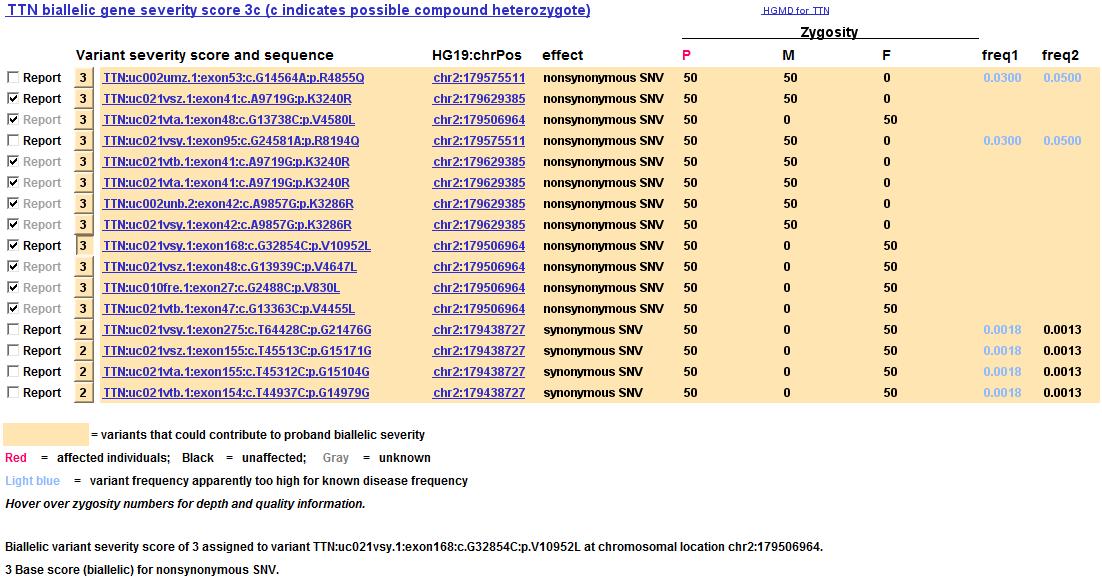
Frequency context for variants (6 June 2016)
Variant frequencies that seem too high to account for rare diseases are now flagged in light blue on the mini variant table display for a gene. In the example below, some TTN variants appear to be too frequent in comparison to known TTN diseases, which have a combined incidence estimated as ~2 per million. This would suggest maximal variant frequencies of 0.0014, so any higher variant frequency is flagged in light blue. Incidence estimates can of course be wrong, so all that is done is flagging, not filtering. But in the example below, variants that are more rare (with no reported frequencies) are the ones chosen to report.
Similar flagging is done on the display for incidental findings. For example, CFTR biallelic diseases are ~326 per million, so any variant with frequency > 0.018 would be flagged. There is no such flagging in the Gene Discovery display since by definition there are no diseases known to be associated with that zygosity of that gene.
"Rule-in, rule-out" mode (1 June 2016)
By default, the display of useful findings represents usefulness of findings in changing the differential diagnosis as a whole. There is now an option in the "Advanced mode" to focus on ruling in or ruling out a particular disease in the differential diagnosis. Simply click on the disease to select it, and then in the "Add findings" and "Add tests" tabs you can now select the option "Rule in or out selected disease", as illustrated in the image below.

Change in severity score range (21 April 2016)
The severity scores now range from 1 (benign, not displayed) to 5 (pathogenic) to correspond to the values used in ACMG guidelines. The analysis is otherwise unchanged; this is just a change in labeling within the software. Previously, values were one number lower. The HTML and XML reports do not use these numbers and are unchanged.
Suppression of noise from over-calling variants (2 March 2016)
As it becomes more common to use whole genome techniques and call intronic variants, some variant tables have included so many variants that some diseases with multiple causative genes were being elevated in the differential diagnosis artifactually. The analysis now uses the zygosity in a disease with most influence on the differential diagnosis and does not double count using other possible zygosities. Notwithstanding, all called zygosities are considered for pathogenicity.
Genome assembly is specifiable in the variant table (23 February 2016)
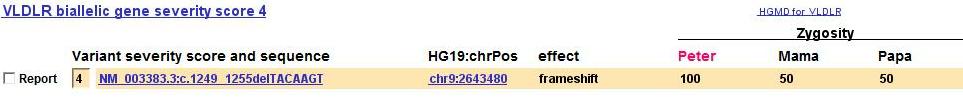
The header in the variant table for the chromosome position is used to specify the genome assembly version used; e.g., HG19:chrPos or HG38:chrPos. The assembly version is displayed in the mini variant table displayed for a gene, and the UCSC genome browser uses the specified assembly version.
Mouse Genome Informatics button added (15 January 2016)
The Gene Discovery page now includes an MGI button to display the Mouse Genome Informatics page about each gene.
Compound heterozygotes from multiple reads in one line (12 October 2015)
Up to 6 non-reference reads with zygosities such as 1/3 and 2/6 can be used in the same row of a variant table, with the same chromosomal location, and compound heterozygotes can be built from these, with labels parsed from alternate sequence fields such as TTG,TT,TTGT,TTGTGTG,T,TTGTG.
Enhanced support for X-linked diseases (16 June 2015)
The SimulConsult clinical diagnostic software and the SimulConsult Genome-Phenome Analyzer are no longer restricted to considering each X-linked disease as being purely X-linked dominant or purely X-linked recessive. Instead, each X-lined disease is annotated in the SimulConsult database to indicate its position on the continuum between recessive and dominant.
For X-linked diseases in which there is a mild female phenotype, the disease is taken seriously whether or not you designate the mother as being affected. The weighting of diseases depends on the degree to which a disease is considered dominant versus recessive.
For "gene discovery" situations in which there is no known human phenotype, both the recessive and dominant zygosities are modeled. In contrast to autosomal diseases, where the zygosity terms monoallelic and biallelic are used, X-linked zygosities are labeled as "one X allele" and "all X alleles" to take into account males having only one X allele. For pseudoautosomal genes, the designations "one XY allele" and "all XY alleles" are used to signal the differences from autosomal inheritance and typical (non-pseudoautosomal) X-linked inheritance.
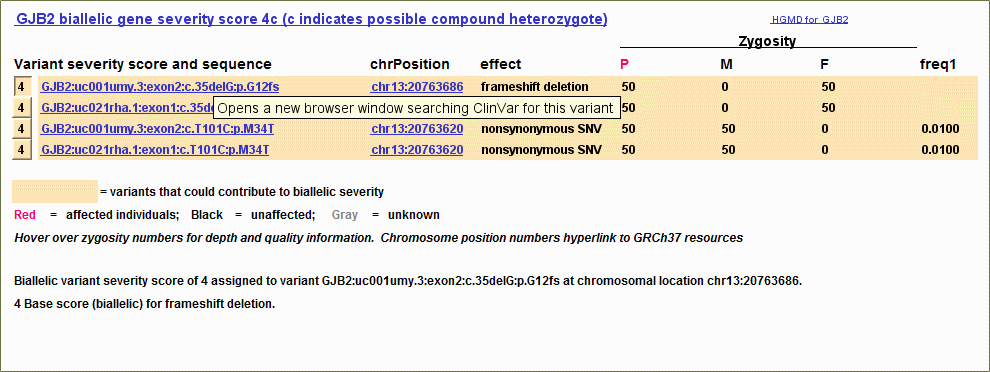
ClinVar links added to variant display (February 2015)
The variant descriptions in the "Variant severity score and sequence" column are now hyperlinked to ClinVar, searching for the gene and the DNA variant. In addition the title of the variant display is hyperlinked to ClinVar, searching for the gene as a whole.
These links are helpful in examining variants on the Genotype tab and on the forthcoming Incidental Findings screen.
Variants are checked for match with stated gender (19 November 2014)
When the software reads a variant table, it checks whether the variants are consistent with the stated gender. The purpose is to detect errors (sample mix-up, incorrect entry of gender) and unusual situations (mosaicism, phenotypic gender not matching chromosomes).
Warnings are flagged in the following circumstances:
- Males with heterozygous calls on the X chromosome that are not pseudoautosomal genes
- Female with calls on the Y chromosome
Errors appear in the "Variant table processing" text area on the File page as the variant file is read. They are of the format: "Proband variants inconsistent with gender in 130 instances".
Assess Disease screen now displays only genes related to that disease (2 June 2014)
Previously, the Assess Disease screen displayed all the genes flagged in the genome-phenome analysis, but this cluttered up the display and made it difficult to see certain findings in the disease. Now, only genes related to the disease being assessed are shown.
Using the maximum of frequency values from the two frequency columns (16 May 2014)
The ability to toggle between using frequency values from freq1 and freq2 columns has been augmented by a third option to use for each variant the higher of the two frequencies. This allows you to use both 1000 Genome and subpopulation frequencies to downplay any variant that is frequent in either set of frequencies.
This option is specified on the "Set variant parameters" screen, using the radio buttons as shown below.
![]()
The default setting is to use column 1, so as to preserve backward compatibility of previous analyses. However, each group can customize defaults for this and other features.
Vigilance for the possibility of tandem repeats (15 May 2014)
By default the software now alerts for the possibility of diseases in which biallelic tandem repeats are pathogenic. It does so when one allele is found to be abnormal, and does so since the other allele being a tandem repeat could result in disease and tandem repeats are not well read on next generation sequencing.
In the example below, the variant table had a monoallelic CSTB variant of severity 4, and the software displays the uncertainty using a question mark instead of a check in commenting on the presence of biallelic variants:

This feature can be toggled off using the checkbox labeled "Call monoallelics in biallelic tandem diseases" on the "Set variant parameters" screen.
Support for old HGNC symbols back to 1 January 2010 (27 February 2014)
As long as the variant table was annotated in 2010 or afterwards, SimulConsult will recognize old HGNC symbols used. The software will refer to the gene by its current HGNC symbol, except when showing the variants, when it will refer to the symbol used, but add the current symbol in parentheses:

Rationale for variant severity score (20 December 2013)
When you examine a list of the variants in each gene, you can now see the rationale for the variant severity score that was assigned. In the figure below, the user has selected the variant severity score 3 button on the left:
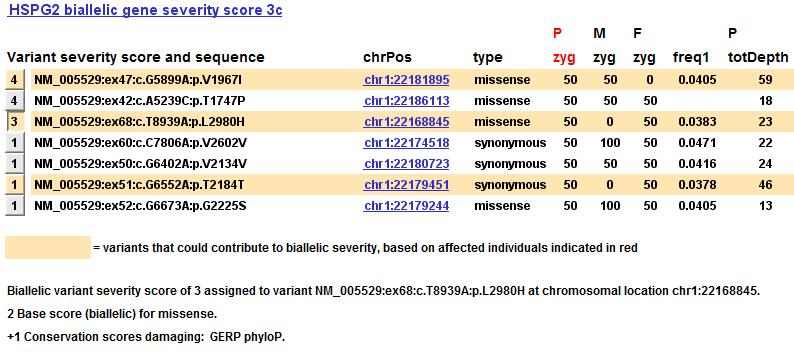
The software displays the various factors that went into that variant severity score, beginning with a score based on the variant type, and changed as appropriate by other scores (functional, conservation, splice, known pathogenicity from databases such as HGMD, homozygous and heterozygous shares, and novelty).